Download Ubuntu 14.04 Virtual Machine Image: here (Please use virtual machine hardware version 11)
Download Windows 8 Virtual Machine Image: here (Please use virtual machine hardware version 11)
(Username: user Password: UserPd@2014)
(Please load your solution in a virtual machine with detailed instructions and then submit it through the above button)
FAQ
Minimum requirement: secure for semi-honest adversaries
Task 1:
Secure Outsourcing GWAS
- Each participating team will be given the genotypes in two groups (one case and one control) of individuals over a few SNPs.
- They are required to develop a
HME based protocol to encrypt these input datasets. Then, the encrypted datasets can be used to compute the minor allele frequencies (MAF) and calculate chi-squared statistics for each of the given SNPs between the case and the control groups on an untrusted remote sever. Finally, the protocol returns the encrypted results (e.g., MAF, chi-squared statistics), where only the data owner with the private key can decrypt the results.
- Each team should submit a suite of programs to implement their algorithms (either binary executable files or Source codes), where the performance will be evaluated by the organizers on a large dataset (consisting of all SNPs within these two groups).
- Download data here:
case group and
control group.
Task 2: Secure comparison between genomic data
- Each participating team will be given two genomic datasets from two individuals, which are organized as the genotypes over many SNPs across the whole human genome.
- They are required to develop a
HME based cryptographic protocol to encrypt both input data and to compute
Hamming distance OR Edit distance between these two encrypted datasets.
- Each team should submit a suite of programs that implements their
algorithms (either
binary executable files or source codes), for which the performance will be evaluated by the organizers on a large dataset (consisting of more pairs of individuals).
- Each team should also provide the security guarantee of their cryptographic protocol.
- Download data here: two sample sequences.
- 100K larger testing dataset is available for downloading here
Minimum requirement: secure for semi-honest adversaries
Task
1: Secure distributed GWAS
- Each participating team will be given the genotypes in two groups (one case and one control) of individuals over a few SNPs. The genotypes of each group will be horizontally partitioned into
two sub-datasets, where each institution will host a single sub-dataset, and cannot directly exchange its dataset with other institutions.
- Participating teams are required to develop a distributed cryptographic protocol to securely aggregate the minor allele frequencies
(MAF) and securely calculate chi-squared statistics for each of the given SNPs between the case and the control groups across multiple institutions.
- Each team should submit a suite of programs to implement their algorithms (either binary executable files or source codes), where the performance will be evaluated by the organizers on a large dataset (consisting of all SNPs within these two groups across multiple institutions).
- Download data here: case group and control group (The first and the second 100 participants are from the first and the second institutions, respectively).
Task 2: Secure comparison between genomic data
- Each participating team will be given two genomic datasets from two individuals, which are organized as the genotypes over many SNPs across the whole human genome.
- They are required to develop a
SMC protocol to compute Hamming distance OR Edit
distance between both datasets, which are distributed in two
different institutions.
- Each team should submit a suite of programs that implements their
algorithms (either
Binary executable files or Source codes), for which the performance will be evaluated by the organizers on a large dataset (consisting of more pairs of individuals).
- Each team should also provide the security guarantee of their cryptographic protocol.
- Download data here: a sequence from institution 1 and a sequence from institution 2. (These files are in VCF (http://samtools.github.io/hts-specs/VCFv4.2.pdf) format.)
- 100K larger testing dataset is available for downloading here
- GWAS (Genome-wide association studies) is a study of many common genetic variants in different individuals (in case-control groups) to see if any variant is associated with a trait (e.g., cancer).
- SNP (Single Nucleotide Polymorphism) is a DNA sequence variation, where a single nucleotide (i.e., A, T, C or G) in the genome differs between individuals.
An example of genotype ‘rs11686243’ of 5 individuals
1 2 3 4 5
rs11686243: AA AG AA AG GG
|
Genotype |
#
of Individuals |
Genotypic
frequencies |
|
AA |
a
= 2 |
AA = 2/5 |
|
AG |
b
= 2 |
AG= 2/5 |
|
GG |
c
= 1 |
GG = 1/5 |
|
Total |
a+b+c
= 5 |
|
SNP
in rs11686243 |
Allele
count |
Allele
frequencies |
Minor
allele? |
|
A |
a*2+b
= 6 |
A = 6/10 |
No |
|
G |
b+c*2
= 4 |
G= 4/10 |
Yes |
Minor allele frequency: 4/10
Association test in
case-control study:
• The chi-squared test is used to determine whether there is a significant difference between the expected frequencies and the observed frequencies
Observed allele counts for SNP1
|
SNP1 |
A |
T |
Total |
|
Case |
a = 3 |
b = 1 |
r = 4 |
|
Control |
c = 1 |
d = 3 |
s = 4 |
|
Total |
a + c |
b+d |
n = 8 |
Expected allele counts for SNP1
|
A |
T |
|
(a+c)*r/n |
(b+d)*r/n |
|
(a+c)*s/n |
(b+d)*s/n |
Chi-squared test
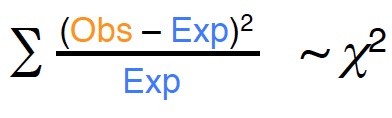
Sequence comparison:
Given two records, x and y, at the same locus from two samples, we may define hamming distance, d, like this: a Python example can be downloaded from here
d = 0;
for all records in the VCF files, which have SVTYPE = SNP or SUB: if given a chrom and pos, there is only one record in one of the VCF file (e.g., x != null), then we set the other record as NULL (e.g., y == null)
if (x == null) || (y == null) || (x.ref == y.ref && x.alt != y.alt)
d += 1;
end for
Edit distance:
Edit distance is known to be hard to calculate in a
privacy-preserving way across two extremely long sequences. To simplify
this computation and still largely keep its accuracy across the whole
genome, we come up with a new definition (hyperlink) that takes
advantage of the unique feature of human genome sequences, which come
with a well-notated genetic variations with regards to the reference
genome (typically stored in Variation Call Format or VCF). Leveraging
their known distances with the reference, we can efficiently approximate
the distance between two genome sequences. Here are the details.
Scalable Edit Distance Approximation for Human Genome
Haixu Tang, XiaoFeng Wang (IUB), Xiaoqian Jiang, Shuang Wang and Lucila
Ohno-Machado (UCSD)
Replacement: calculate just like Hamming distance
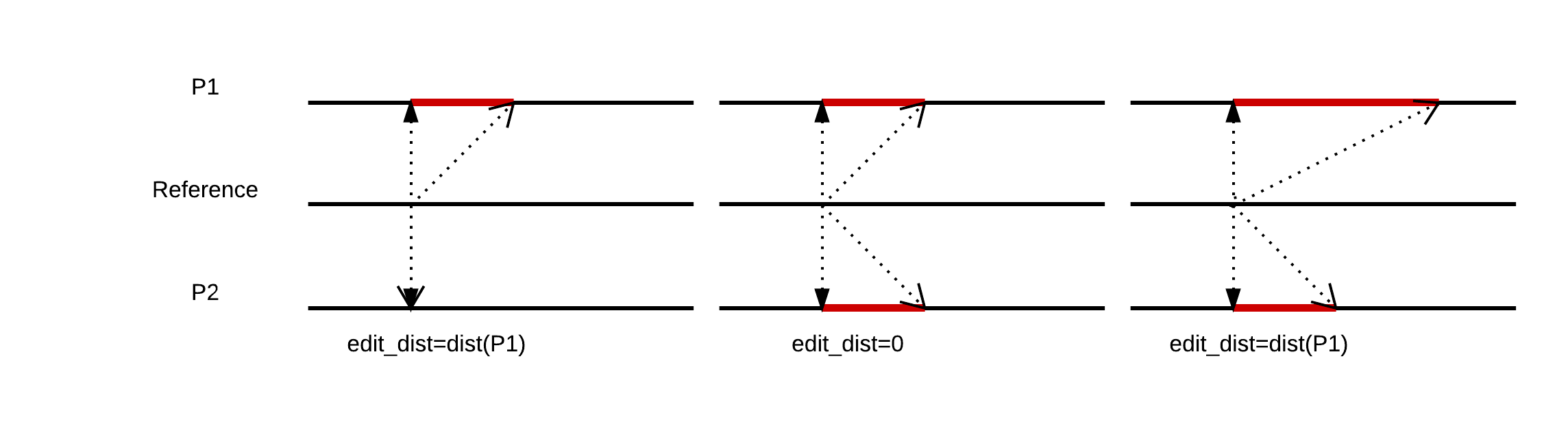
Insertion/deletion: if P1 includes an insertion or deletion compared
with the reference while P2 does not, use P1’s distance as the edit
distance for the current locus and then realign P1 and P2 to skip the
inserted or deleted subsequence.
If both P1 and P2 have insertion or deletion at the same locus, choose
the larger one as the approximation for the edit distance at the locus,
and realign P1 and P2.
The following figure shows an example.
For example: given two records, x and y, at the same locus from two samples (the record is empty if there is no variation in one sample, len(empty)=0), we may define edit distance, d, like this: a Python example can be downloaded from here
d = 0;
for all records in the VCF files:
1. if x == y, continue;
2. if x != y, d += max(D(x), D(y))
end for
where D(x):
if x.svtype == snp, D(x) = 1
if x.svtype == sub, D(x) = len(x)
if x.svtype == ins, D(x) = len(x)
if x.svtype == del, D(x) = len(x)
